Las enzimas son moléculas intracelulares que aceleran y dirigen la conversión de un metabolito a otro, siendo así la piedra angular del metabolismo del cuerpo. No es de extrañar, pues, que nuestro organismo cuente con más de 75.000 enzimas diferentes, estando cada una de ellas especializada en una función muy concreta.
Y, como bien sabemos, la síntesis de todas estas enzimas está codificada en nuestros genes, las unidades de ADN donde está escrita la información necesaria para regular nuestra fisiología. Y estos genes, lejos de ser unidades indestructibles, pueden sufrir errores o mutaciones.
Y en este sentido, ¿qué sucede cuando la mutación genética se produce en un gen que codifica para alguna de las enzimas que hemos visto? Pues, básicamente, que se abre la puerta a que desarrollemos una enfermedad a causa de esta carencia enzimática.
Hoy hablaremos de uno de estos trastornos: la fenilcetonuria. Una enfermedad genética y hereditaria en la que, debido a la ausencia de la enzima degradadora de la fenilalanina, este aminoácido presente en alimentos proteicos se acumula de forma peligrosa en nuestro organismo. Veamos las causas, síntomas y tratamiento de esta patología.
- Te recomendamos leer: “Las 15 enfermedades genéticas más comunes: causas, síntomas y tratamiento”
¿Qué es la fenilcetonuria?
La fenilcetonuria es una enfermedad genética y hereditaria cuyos síntomas se deben a la acumulación de la fenilalanina, un aminoácido presente en alimentos proteicos, en el organismo de forma especialmente peligrosa en sangre y cerebro. Se trata de un trastorno poco frecuente en el que la persona nace con una mutación genética que le impide sintetizar la enzima que degrada este aminoácido.
La fenilalanina es uno de los 9 aminoácidos esenciales, lo que significa que solo pueden obtenerse a través de la alimentación. Es imprescindible para el correcto desarrollo y funcionamiento neuronal, pues las proteínas que se obtienen a partir de ella regulan la síntesis de endorfinas, reducen la experimentación de dolor y de apetito, controlan la producción de adrenalina y dopamina y generan estrés pero estimulando también la memoria, la vitalidad y el aprendizaje.
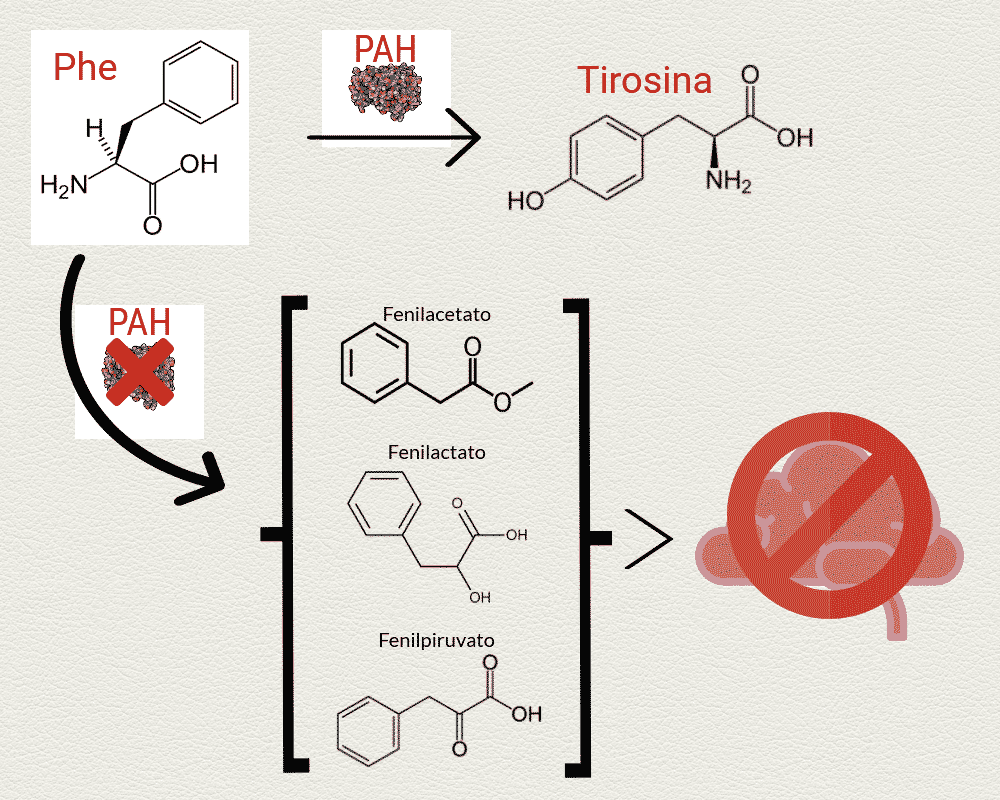
Pero para obtener estas proteínas, la fenilalanina obtenida a través del consumo de alimentos proteicos debe procesarse. Y aquí entra en juego la fenilalanina hidroxilasa, una enzima que actúa a nivel de hígado y que se encarga de degradar la fenilalanina y convertirla en tirosina, la cual sigue la ruta de síntesis de proteínas.
Las personas con fenilcetonuria, debido a una mutación de un gen localizado en el cromosoma 12, son incapaces de producir la enzima fenilalanina hidroxilasa, dando lugar así a una alteración metabólica congénita en la que el aminoácido fenilalanina, al no poder degradarse en tirosina y continuar la ruta del metabolismo proteico, se acumula inevitablemente en el cuerpo.
Esta acumulación, además de hacer que la persona sea de piel muy clara y ojos azules (luego veremos por qué), provoca daños en todo el cuerpo, incluidos fenómenos de discapacidad intelectual y trastornos psicológicos debido al efecto tóxico que tiene la fenilalanina acumulada (e imposible de procesar y de eliminar del organismo) en el cerebro.
Se trata de una enfermedad rara con una incidencia de aproximadamente 1 caso por cada 10.000 nacimientos, pero aun así es importante entender su naturaleza ya que no existe cura y el único tratamiento posible es seguir, durante toda la vida, una dieta lo más pobre posible en proteínas. Es decir, lo único que puede hacerse es evitar que la fenilalanina, que no podrá ser degradada, entre en el cuerpo.
- Te recomendamos leer: “Los 20 aminoácidos (esenciales y no esenciales): características y funciones”
Causas
La fenilcetonuria es una enfermedad genética rara con una incidencia de 1 caso por cada 10.000 nacimientos que, como hemos visto, tiene una causa clara: la ausencia de la fenilalanina hidroxilasa, la enzima que degrada el aminoácido fenilalanina.
Pero, ¿qué es lo que hace que una persona no disponga de esta enzima? Básicamente, una mutación genética con un claro factor hereditario. La fenilcetonuria sigue un patrón de herencia genética de tipo autosómico recesivo.
La mutación que da lugar a la fenilcetonuria está localizada en el gen PAH (locus 12q22-q24.2), el cual está presente en el cromosoma 12. Dependiendo de qué forma esté alterada la secuencia genética, la síntesis de la enzima estará más o menos dañada y, por lo tanto, la fenilcetonuria será leve, moderada o grave.
Aun así, hay que tener en cuenta que es una mutación recesiva. Los seres humanos tenemos 23 pares de cromosomas, lo que significa que tenemos dos copias de cada cromosoma. Y, en este sentido, tenemos dos copias del gen PAH ya que hay dos cromosomas 12.
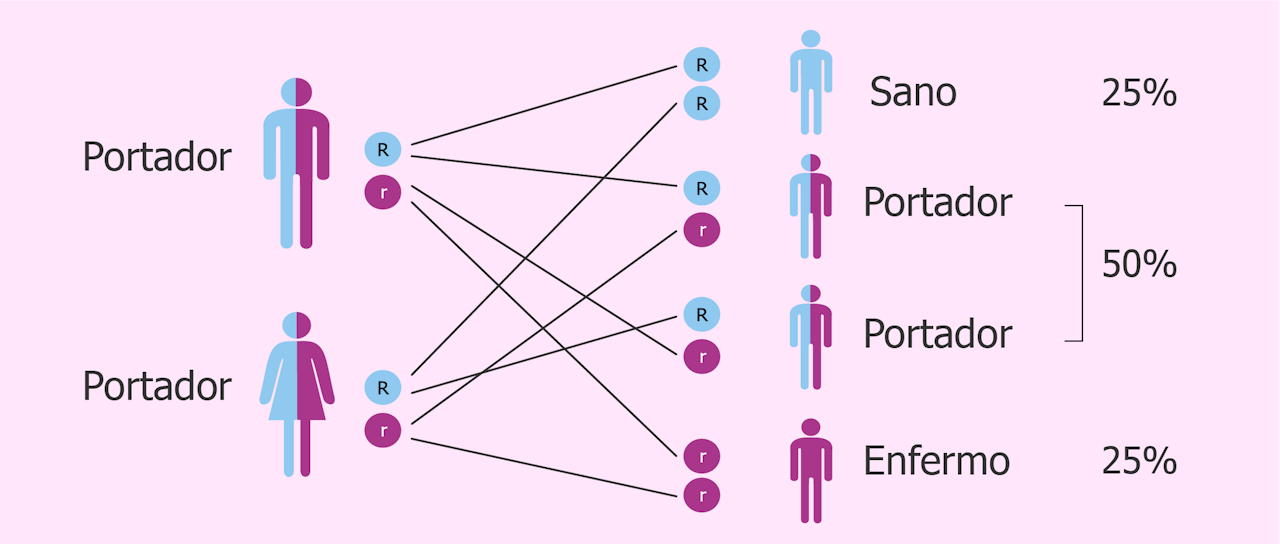
¿Qué pasa si solo uno de los dos genes PAH está mutado? Simplemente, nada. La persona es portadora de la mutación que conduce a la fenilcetonuria, pero tiene un gen sano que contrarresta dicha mutación, por lo que puede sintetizar la enzima fenilalanina hidroxilasa y, por lo tanto, no sufrirá nunca la enfermedad.
La fenilcetonuria solo se expresa cuando la persona dispone de los dos genes PAH mutados. Por lo tanto, si ponemos que, por ejemplo, el padre es portador de la mutación (solo tiene un gen mutado) pero la madre no es ni siquiera portadora (ningún gen mutado), el riesgo de que sus hijos sufran fenilcetonuria es del 0%.
Ahora bien, si, por ejemplo, tanto el padre como la madre son portadores (tienen uno de los dos genes mutados), la probabilidad de que su hijo sufra la enfermedad (herede justo los dos genes mutados) es del 25%. En esto se basa la herencia autosómica recesiva. Los dos padres tienen que tener, al menos, un gen defectuoso para que su hijo desarrolle la enfermedad.
Esto explica que su incidencia sea baja, de 1 caso por cada 10.000 nacimientos. Aun así, se cree que el 2% de la población podría ser portadora de la enfermedad, en el sentido que tienen uno de los dos genes que codifica para la enzima, mutado. Curiosamente, esta frecuencia varía entre grupos étnicos y se ha visto que la población afroamericana es menos portadora de esta mutación.
- Te recomendamos leer: “Los 11 tipos de mutaciones (y sus características)”
Síntomas
La persona nace con la enfermedad, pero al principio de la vida, la fenilcetonuria no da señales de su presencia ya que todavía no ha habido tiempo para que la acumulación de fenilalanina pase el umbral del peligro y provoque síntomas.
Dependiendo tanto del nivel de afectación en la síntesis de la enzima (no siempre hay una ausencia total de la fenilalanina hidroxilasa) como del estilo de vida de la persona (del consumo que haga de alimentos proteicos), la acumulación de fenilalanina en el organismo provocará signos clínicos antes o después y lo hará de forma más o menos grave.
Sea como sea, los principales síntomas son los siguientes: piel y ojos muy claros (la persona no puede producir melanina de forma normal ya que la degradación de la fenilalanina es parte importante de su síntesis), erupciones cutáneas, temblores, espasmos en extremidades, hiperactividad, microcefalia (cabeza anormalmente pequeña), convulsiones, olor extraño (similar a humedad o a moho) en piel, orina y aliento, retrasos en el desarrollo, problemas de comportamiento, alteraciones emocionales, dificultades para socializar, trastornos psiquiátricos y, en caso de estar embarazada, sufrir la patología y no tratarla, problemas en el desarrollo fetal (bajo peso al nacer, defectos cardíacos, anomalías faciales, discapacidad intelectual…).
Como vemos, la acumulación de fenilalanina en el organismo puede ser muy peligrosa y, además, es irreversible. No se puede eliminar del cuerpo la que ya está acumulada y, en caso de seguir introduciéndola, el problema solo hará que ir a más.
Y es aquí cuando se abre la puerta a las complicaciones asociadas. Si no se aborda clínicamente desde el nacimiento, la fenilcetonuria puede provocar problemas neurológicos graves, daños en la salud cardiovascular potencialmente fatales, graves problemas de conducta y daños cerebrales irreversibles. Aun así, pese a que no tenga cura, la fenilcetonuria puede (y debe) tratarse. Veamos cómo.
- Te recomendamos leer: “Los 24 síntomas en bebés que deben alertarte”
Tratamiento
La fenilcetonuria es una enfermedad irreversible e incurable (como sucede con todos los trastornos genéticos), pero esto no significa que no pueda tratarse. Un simple análisis de sangre en bebés que presentan las señales que hemos comentado es suficiente para diagnosticar la fenilcetonuria. Y a partir de ese momento, el tratamiento debe empezar cuanto antes.
El tratamiento es muy sencillo de comprender pero muy difícil de poner en práctica: seguir, de por vida, una dieta muy limitada en proteínas. Como hemos dicho, la fenilalanina está presente en todos los alimentos proteicos (carne, pescado, leche, huevos, legumbres, nueces, queso, soja, frijoles…), por lo que, teniendo en cuenta de que no hay forma de recuperar la actividad de la enzima que la degrada ni de revertir la acumulación, la única forma de abordar la enfermedad es comer cuanta menos proteína posible a lo largo de toda la vida.
La idea del tratamiento es ver hasta qué punto la actividad de la enzima está dañada para desarrollar una dieta donde se introduce la suficiente fenilalanina para el correcto desarrollo fisiológico pero sin sobrepasar el umbral después del cual la acumulación resultará demasiado tóxica. Esta ingesta prudente de fenilalanina podrá ir cambiando a lo largo de la vida, así que habrá que hacer revisiones periódicas.
Además de los evidentes productos proteicos cuyo consumo tendrá que reducirse al máximo, también habrá que evitar los alimentos que contengan aspartamo (que es un edulcorante artificial hecho con la fenilalanina) e incluso puede que tenga que limitarse la ingesta de cereales y patatas.
De todas formas, ante el diagnóstico de fenilcetonuria, el médico pondrá al bebé y a la familia en manos de un nutricionista que desarrollará una dieta para que el efecto de la enfermedad en el presente y futuro de la persona sea mínimo. Si esta dieta se introduce precozmente a las pocas semanas de vida, el riesgo de las complicaciones neurológicas más graves será mínimo y el pronóstico, muy bueno.